Cluster AL Fitting Mode
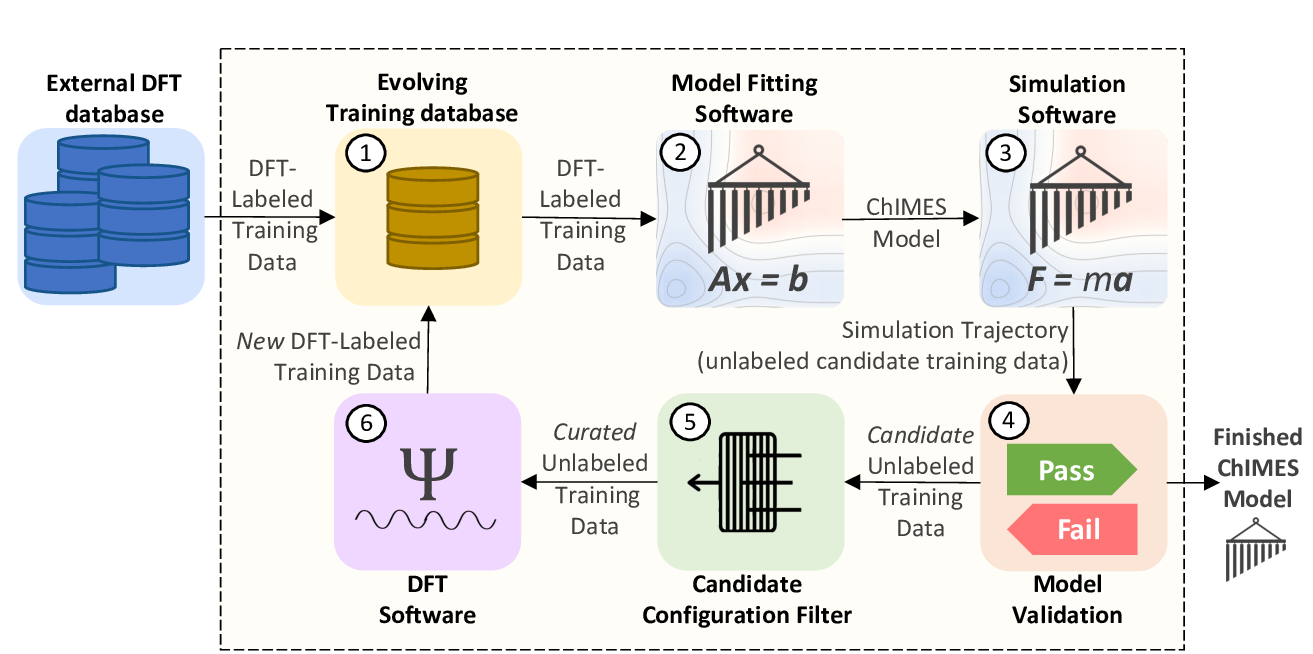
Fig. 1: The ChIMES Active Learning Driver Workflow.
When developing models for molecular reacting systems, our cluster-based active learning (AL) can be advantageous. This AL strategy attemps to improve description of conformational energetics and nominal reaction barriers. This is achieved by supplementing the basic mode by carving out candidate molecules and nominal transition states from DFT- and ChIMES-generated simulation trajectories , down-selecting a maximally informative subset, and adding them to the training set. Details of this strategy are outlined in R.K. Lindsey et al, JCP 2020.
… Warning:
This capability will only run on Slurm systems, and may require a specific Slurm specification. See the ``utilities/new*sh`` files for details.
Example Fit: Water
Note
Files for this example are located in ./<al_driver base folder>/examples/cluster_based_active_learning_single_statepoint-VASP/
In this section, an example 1-iteration fit for water at 1000 K and 1.25 g/cc is overviewed. The model will include up-to-three body interactions with the following hyperparameters. Note: This example is intended to run quickly and will not yield neither a quality nor stable model.
Hyperparameter |
Value |
|---|---|
2-body order |
12 |
2-body outer cutoff |
5 |
3-body order |
3 |
3-body outer cutoff |
3.5 |
OO inner cutoff |
2.31 |
HH inner cutoff |
1.18 |
OH inner cutoff |
0.82 |
Morse lambda |
1.00 |
Tersoff parameter |
0.75 |
Input Files
The neccesary input files and directory tree structure are provided in the example folder, i.e.:
$: tree
.
├── ALL_BASE_FILES
│ ├── ALC-0_BASEFILES
│ │ ├── fm_setup.in
│ │ ├── reactive_water.temps
│ │ ├── reactive_water.xyzf
│ │ └── traj_list.dat
│ ├── CHIMESMD_BASEFILES
│ │ ├── bonds.dat
│ │ ├── case-0.indep-0.input.xyz
│ │ ├── case-0.indep-0.run_md.in
│ │ └── run_molanal.sh
│ ├── QM_BASEFILES
│ │ ├── 1000.INCAR
│ │ ├── H.POTCAR
│ │ ├── KPOINTS
│ │ └── O.POTCAR
│ ├── run_md.cluster
│ ├── loose_bond_crit.dat
│ └── tight_bond_crit.dat
Beginning with the contents of the ALC-0_BASEFILES folder: fm_setup.in, traj_list.dat, and the training trajectory (reactive_water.xyzf) require no special treatment for cluster-based AL. However, an additional file (reactive_water.temps) is now required. This file must have the same name as the training .xyzf file and end with a “.temps” extension. For each frame in the .xyzf file, the .temps file contains the corresponding target system temperature.
The contents of the CHIMESMD_BASEFILES and QM_BASEFILES foldera also requires no special treatment.
Three new files are required, which sit directly in the ALL_BASE_FILES folder: run_md.cluster, tight_bond_crit.dat, and loose_bond_crit.dat. The run_md.cluster file can be taken exactly as provided in the example folder. The tight* and loose* files provide the bonding distanct criteria used to identify molecules and nominal transition state species, respctively. The format of each file is as follows: The first line gives a space-separated list of each element present in the system (e.g., “O H”). The second line gives the unique number of atom pair types formed by those atoms, e.g., O an H can form 3 pairs, O O, O H, and H H. Then, one line is given for each pair, which gives the two atom types and the corresponding distance criteria, (e.g., “H H 1.4”).
Contents of the config.py file must be modified to reflect your HPC system and absolute paths prior to running this example. File contents specific to/required for cluster-based AL are highlighted below:
1################################
2##### General options
3################################
4
5ATOM_TYPES = ['O', 'H']
6NO_CASES = 1
7
8DRIVER_DIR = "/p/lustre3/lindsey11/al_driver-myLLfork/"
9WORKING_DIR = "/p/lustre3/lindsey11/al_driver-myLLfork/examples/cluster_based_active_learning_single_statepoint-VASP/"
10CHIMES_SRCDIR = "/p/lustre3/lindsey11/test_chimes_lsq-for-LL_to_ext_PR/chimes_lsq-LLfork/src/"
11
12################################
13##### General HPC options
14################################
15
16HPC_ACCOUNT = "iap"
17HPC_PYTHON = "/usr/tce/bin/python3"
18HPC_SYSTEM = "slurm"
19HPC_PPN = 56
20
21HPC_EMAIL = False
22
23################################
24##### ChIMES LSQ
25################################
26
27ALC0_FILES = WORKING_DIR + "ALL_BASE_FILES/ALC-0_BASEFILES/"
28CHIMES_LSQ = CHIMES_SRCDIR + "../build/chimes_lsq"
29CHIMES_SOLVER = CHIMES_SRCDIR + "../build/chimes_lsq.py"
30CHIMES_POSTPRC= CHIMES_SRCDIR + "../build/post_proc_chimes_lsq.py"
31
32# Generic weight settings
33
34WEIGHTS_FORCE = [ ["A"], [[1.0 ]] ]
35WEIGHTS_FGAS = [ ["A"], [[1.0 ]] ]
36WEIGHTS_ENER = [ ["A"], [[0.3 ]] ]
37WEIGHTS_EGAS = [ ["A"], [[1.0 ]] ]
38WEIGHTS_STRES = [ ["A"], [[100.0]] ]
39
40REGRESS_ALG = "dlasso"
41REGRESS_VAR = "1.0E-5"
42REGRESS_NRM = True
43
44# Stress tensor settings
45
46STRS_STYLE = "ALL" # Options: "DIAG" or "ALL"
47
48CHIMES_BUILD_NODES = 1
49CHIMES_BUILD_QUEUE = "pdebug"
50CHIMES_BUILD_TIME = "01:00:00"
51
52CHIMES_SOLVE_NODES = 2
53CHIMES_SOLVE_QUEUE = "pdebug"
54CHIMES_SOLVE_TIME = "01:00:00"
55
56################################
57##### Do Cluster-based active learning
58################################
59
60DO_CLUSTER = True
61MAX_CLUATM = 10
62TIGHT_CRIT = WORKING_DIR + "ALL_BASE_FILES/tight_bond_crit.dat"
63LOOSE_CRIT = WORKING_DIR + "ALL_BASE_FILES/loose_bond_crit.dat"
64CLU_CODE = DRIVER_DIR + "/utilities/new_ts_clu.cpp"
65
66MEM_BINS = 40
67MEM_CYCL = MEM_BINS/10
68MEM_NSEL = 100
69MEM_ECUT = 4000.0
70
71CALC_REPO_ENER_CENT_QUEUE = "pdebug"
72CALC_REPO_ENER_CENT_TIME = "1:00:00"
73
74CALC_REPO_ENER_QUEUE = "pdebug"
75CALC_REPO_ENER_TIME = "1:00:00"
76
77
78################################
79##### Molecular Dynamics
80################################
81
82MD_STYLE = "CHIMES"
83CHIMES_MD_MPI = CHIMES_SRCDIR + "../build/chimes_md"
84CHIMES_MD_SER = CHIMES_SRCDIR + "../build/chimes_md-serial"
85
86MOLANAL = CHIMES_SRCDIR + "../contrib/molanal/src/"
87MOLANAL_SPECIES = ["H2O","H3O", "OH"]
88
89MD_NODES = [1] * NO_CASES
90MD_QUEUE = ['pdebug'] * NO_CASES
91MD_TIME = ['00:05:00'] * NO_CASES
92
93################################
94##### QM-Specific variables (Single point calculations)
95################################
96
97QM_FILES = WORKING_DIR + "ALL_BASE_FILES/QM_BASEFILES"
98
99VASP_EXE = "/p/lustre3/lindsey11/vasp_std.5.4.4"
100VASP_TIME = "01:00:00"
101VASP_NODES = 1
102VASP_PPN = 56
103VASP_QUEUE = "pdebug"
104VASP_MODULES = "intel-classic/19.1.2 mvapich2/2.3.6 mkl"
The variable DO_CLUSTER controls whether cluster-based AL is used. This variable is false by default; when false, no variables in the “Do Cluster-based active learning” block above need be specified. MAX_CLUATM controls the maximum number of atoms that a molecule can be comprised of. TIGHT_CRIT and LOOSE_CRIT are the full paths to the tight and loose bond criteria files in the ALL_BASE_FILES folder. CLU_CODE is the path to the cluster-extraction code.
The next chunk of variables control the cluster down-selection process. MEM_BINS is the number of bins in the cluster energy histogram, MEM_CYCLE is the number of Monte Carlo cycles to perform during the down-selection process, MEM_NSEL is the number of molecules to select each AL cycle, and MEM_ECUT a cutoff the ignores any molecules whose absolute energy is greater than MEM_ECUT.
CALC_REPO_ENER_CENT* and CALC_REPO_ENER specify computational resources for assinging ChIMES energies to each candidate cluster.